Blodsyndrom: Symptom, orsaker och behandling
Blodsyndrom (BS) är en sällsynt sjukdom av autosomalt recessivt arv som huvudsakligen kännetecknas av tre aspekter: tillväxt retardation, överkänslighet mot solen och telangiektasi i ansiktet (dilation av kapillärkärl). Dessa patienter har en genomisk instabilitet som predisposes dem till att utveckla cancer lätt.
Det upptäcktes av dermatologen David Bloom år 1954 genom observation av flera patienter som presenterade dvärg och telangiectatisk erytem (rodnad hud på grund av utvidgning av blodkarillärer) (Elbendary, 2015).
Detta syndrom kan också kallas medfödd telangiectatisk erytem eller Bloom-Torre-Machacek syndrom.
Orsaker till Bloom syndrom
Bloom syndromet är en autosomal recessiv sjukdom, det vill säga en mutation måste inträffa i båda alleler av BLM-genen, både från moderen och från fadern (Ellis et al., 1995). Föräldrar behöver inte nödvändigtvis ha denna sjukdom, men de kan vara bärare av den muterade genen utan att ha symtom.
Mer än 60 mutationer har hittats i BLM-genen i Blooms syndrom, den vanligaste är deletionen av 6 nukleotider i position 2281 och substitutionen av 7 andra (Elbendary, 2015).
Enligt Genetics Home Reference (2016) är BLM-genen ansvarig för att skicka instruktioner för skapandet av RecQ-proteinet, som ingår i familjen av helikaser.
Vilka helikaser gör det är att ansluta sig till DNA och tillfälligt separera de två strängarna av det, som vanligtvis är kopplade i en spiral, i syfte att utveckla processer som replikering (eller DNA-kopia), förberedelser för celldelning och reparation. av DNA-skada.
Kortfattat är RecQ-helikaser viktiga för att upprätthålla strukturen hos DNA och är därför kända som "genomomsorgare".
Till exempel när en cell ska delas för att bilda två nya celler, måste det DNA som finns i kromosomerna kopieras så att varje ny cell har två kopior av varje kromosom: en av fadern och en annan av mamman.
DNA kopierat från varje kromosom har två identiska strukturer som kallas systerkromatider, och de är länkade i början innan cellernas uppdelning äger rum.
I detta skede byter de några bitar av DNA mellan dem; vad som är känt som systerkromatidutbyte. Det verkar som att denna process förändras i Blooms sjukdom, eftersom BLM-proteinet är skadat och det här kontrollerar de korrekta utbytena mellan systerskromatiderna och att DNA-en förblir stabil vid kopieringstidpunkten.
I själva verket finns det i genomsnitt 10 mer än normala utbyten mellan kromatider i Bloom syndromet (Seki et al., 2006).
Å andra sidan finns det också raster i det genetiska materialet i denna sjukdom, vilket orsakar försämring av normala cellulära aktiviteter som, på grund av bristen på BLM-proteinet, inte kan repareras.
Faktum är att vissa experter klassificerar detta syndrom som "kromosombrottsyndromet", eftersom det är relaterat till ett stort antal raster och omarrangemang av kromosomerna.
Denna instabilitet av kromosomerna medför en större sannolikhet att utveckla sjukdomar. Till exempel, på grund av bristen på BLM-proteinet kan de inte återhämta sig från DNA-skador som kan orsaka ultraviolett ljus och därför är dessa patienter ljuskänsliga.
Dessutom har de drabbade en immunbrist som gör dem mer mottagliga för infektion.
Å andra sidan har de stor sannolikhet att utveckla cancer i något organ genom okontrollerad cellfördelning, som främst framträder leukemi (det är en typ av blodcancer som kännetecknas av ett överskott av vita blodkroppar) och lymfom (cancer i lymfkörteln i systemet). immun).
Fel har också hittats i FANCM-genens verkan, som ansvarar för kodningen av MM1- och MM2-proteiner, som också tjänar till att reparera DNA-skador.
Dessa är de som har kopplats både till detta syndrom och till Fanconi anemi. Det är därför vi ser att dessa två sjukdomar är lika i deras fenotyp och i predispositionen till hematologiska tumörer och insufficiens i benmärgen.
Hur som helst, de molekylära mekanismerna som påverkar kromosomerna i Bloom syndromet är fortfarande undersökta.
Vad är dess förekomst?
Bloom syndromet är relativt sällsynt, endast omkring 300 fall som beskrivs i den medicinska litteraturen är kända. Även om denna sjukdom uppträder hos många etniska grupper, verkar det vara mycket vanligare i Ashkenazi-judarna, som står för 25% av patienterna med detta syndrom.
Faktum är att inom denna etniska grupp kan frekvensen av syndromet uppgå till 1%. Det har också hittats, om än mindre, i japanska familjer.
När det gäller kön verkar män vara något mer benägna att få sjukdomen än kvinnor, där andelen är 1, 3 män för 1 kvinna.
Vad är dess symptom?
Detta tillstånd uppstår redan under de första månaderna av livet och för närvarande har ingen av patienterna levt mer än 50 år.
- Maligna tumörer : orsakad av genomisk instabilitet som förklarats ovan är den främsta orsaken till dödsfall hos de som drabbas av detta syndrom. Enligt den nationella organisationen för sällsynta sjukdomar (2014) kommer omkring 20% av de som drabbas av Bloom syndrom att utveckla cancer. Dessa patienter har mellan 150 och 300 gånger större risk att utveckla cancer än människor utan denna sjukdom.
- Immunbrist som varierar i svårighetsgrad enligt patienten och predisponerar för olika infektioner. Detta uppstår på grund av underskott i proliferation av lymfocyter (vita blodkroppar), problem i syntesen av immunoglobulin (antikroppar av immunsystemet) och lågt respons på stimulering av mitogener (som styr division och tillväxt av celler).
- Defekter i T- och B-lymfocyter är vanliga, vilket påverkar immunsystemets utveckling.
- Felaktigheten i immunförsvaret kan leda till öroninfektion (främst otitis media), lunginflammation eller andra tecken som diarré och kräkningar.
- Fotosensitivitet : Det är en överdriven känslighet av DNA: n för ultravioletta strålar och blir skadad. Det anses vara en form av fototoxicitet eller celldöd som skadar den drabbas hud när solen träffar.
- Minskning av fertilitet eller infertilitet . Faktum är att det hos män inte finns någon förmåga att producera väntar. Hos kvinnor finns det en mycket tidig klimakteriet.
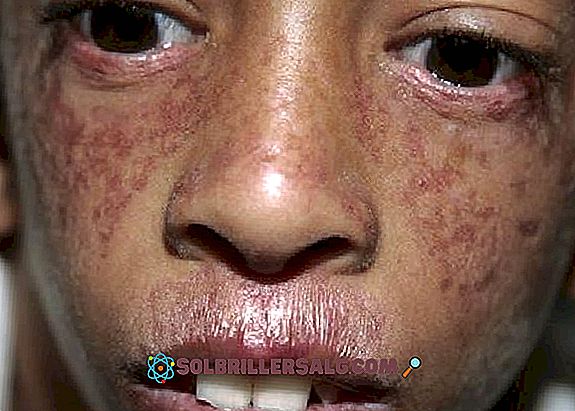
- Kutan manifestationer : Förutom ljuskänslighet uppträder poikiloderma också, en hudpåverkan som uppträder huvudsakligen i nacken, uppträder hypopigmenterade zoner, andra hyperpigmenterade områden, telangiectasier och atrofi. Det är vanligen observerade röda fläckar på huden som är förknippade med sol exponering (särskilt i ansiktet).
- Ett annat kutan problem som observeras är telangiectasi, vilket ses som rödaktiga utbrott i ansiktet som orsakas av utvidgning av små blodkärl. Det verkar som ett "fjäril" mönster som täcker näsan och kinderna.
- Onormala bruna eller gråa fläckar kan också förekomma på andra delar av kroppen ("café au lait" fläckar).
- Fördröjning i utvecklingen som redan uppträder hos spädbarn. De små har vanligtvis ett distinkt huvud och ansikte, smalare och mindre än normalt.
- Omkring 10% av de drabbade slutar utveckla diabetes (National Organization for Rare Disorders, 2014).
- Mycket skarp röst .
- Förändringar i tänderna .
- Onormala ögon, öron (framstående öron), händer eller fötter (som polydaktyly, som uppstår när patienten har fler fingrar än normalt).
- Pilonidala cyster.
- Foderproblem : De märks speciellt hos spädbarn och småbarn, vilket visar brist på intresse för att äta. Det åtföljs många gånger med svår gastroesofageal reflux.
- Den intellektuella förmågan är varierbar, så att de hos vissa patienter blir mer försämrade och i andra är de normala.
Hur diagnostiseras det?
Det kan diagnostiseras av någon av följande test:
- Cytogenetiska tester som mäter kromosomavvikelser och utbytesnivån hos systerskromatider.
Det är möjligt att observera närvaron av fyra-radiala föreningar (utbyte av fyraarmkromatider) i lymfocyter odlade i blod för att kontrollera om det finns höga nivåer av utbyte av systerskromatider i vilken cell som helst, kromatidgap, raster eller omarrangemang; Eller se direkt om det finns mutationer i BLM-genen.
Dessa tester kan upptäcka en frisk individ som bär mutationer i BLM-genen och kan överföra dem till sina avkommor.
Food and Drug Administration of the United States (FDA) tillkännagav i februari 2015 kommersialiseringen av ett genetiskt test av "23andMe" som kan vara användbart för att upptäcka tidigt förekomsten av denna sjukdom.
Förekomsten av detta syndrom bör misstänkas om dessa kliniska tillstånd föreligger:
- Fördröjning i den signifikanta tillväxten sedan intrauterin perioden.
- Förekomst av erytemor på ansiktets hud efter exponering för solen.
Förvirra inte med ...
Följande syndrom måste beaktas för att reglera dem innan de diagnostiserar Blooms syndrom:
- Andra autosomala recessiva kromosomala instabilitetssyndrom som är förknippade med brott och omplaceringar av kromosomer, vilket gör ämnet speciellt utsatt för vissa typer av cancer, såsom: Fanconi anemi, ataxia telangiectasia eller xeroderma pigmentosa som involverar andra gener och inte BLM .
- Cockayne syndrom, som består av en ärftlig sjukdom som uppträder vid fördröjd utveckling, ljuskänslighet och åldrande utseende i ung ålder. Det är en sällsynt form av dvärg.
- Rothmund-Thomson syndrom : extremt ovanligt och manifesteras av typiska hudavvikelser, hårdefekter, juvenil grå störningar, kortstorhet och skelettförändringar som kraniofaciala missbildningar. Det liknar Blooms syndrom vid hudinflammation, poikiloderma, huddegenerering (atrofi) och telangiektasi.
behandling
Det finns ingen specifik behandling för Bloom syndrom, det vill säga för det alltför stora antalet mutationer. Snarare är insatserna inriktade på att lindra symtom, erbjuda stöd och förebygga komplikationer.
- Försök inte exponera dig själv direkt under solen.
- Använd tillräckligt med solskyddsmedel.
- Uppföljning av en hudläkare, för att behandla fläckar, rodnad och inflammation i huden.
- Använd antibiotika för infektioner.
- Periodiska medicinska kontroller för att upptäcka möjliga fall av cancer, främst när dessa patienter når vuxen ålder. Vi måste försöka vara uppmärksamma på de möjliga symtomen, eftersom det finns tumörer som kräver ett tidigt kirurgiskt avlägsnande för deras återhämtning. Vissa metoder för tidig diagnos av cancer är mammografi, Papanicolaou eller vaginal cytologi eller koloskopi.
- Kontrollera att dessa barn får de nödvändiga näringsämnena som försöker intervenera i matsmältningsrefluxen. För detta kan ett rör placeras i övre delen av tarmkanalen för komplementär utfodring medan du sover. Det kan öka de små fettavlagorna lite, men det verkar inte ha någon effekt på tillväxten själv.
- Undersök förekomsten av diabetes för att behandla den så snart som möjligt.
- Om individen har cancer kan benmärgstransplantation övervägas.
- Familj och andra stödgrupper och föreningar med liknande sjukdomar så att den drabbade personen utvecklas som en person med högsta möjliga livskvalitet.
- Om familjen har haft fall av denna sjukdom eller av makens familj, skulle genetisk rådgivning vara användbar för att få information om arten, arvet och konsekvenserna av denna typ av sjukdomar för att bidra till medicinsk beslutsfattande och personligt.







